
琉球大学 戦略的研究プロジェクトセンターの佐藤行人・特命講師と鶴井(佐藤)香織・特命助教、同大学 農学部の辻瑞樹・教授、立田晴記・教授と加藤三歩・研究員、および同大学院 医学研究科の木村亮介・准教授らの研究チームによる成果が、進化生態学の学術誌「Ecology and Evolution」に掲載されました。
<発表のポイント>
◆少量のDNAサンプルでも、PCR増幅により遺伝的多様性を調べるMIG-seqという手法で、琉球列島の擬態チョウ・シロオビアゲハの遺伝的関係を詳細に推定
◆推定された高精度な遺伝的距離に基づいて、各島の擬態型の進化パターンを解析
◆琉球列島に見られるベイツ型擬態の混在性は、遺伝的な類縁や隔離ではなく、各島の捕食圧に応じて進化したという説を、細胞核の遺伝子解析から裏付けた
<発表概要>
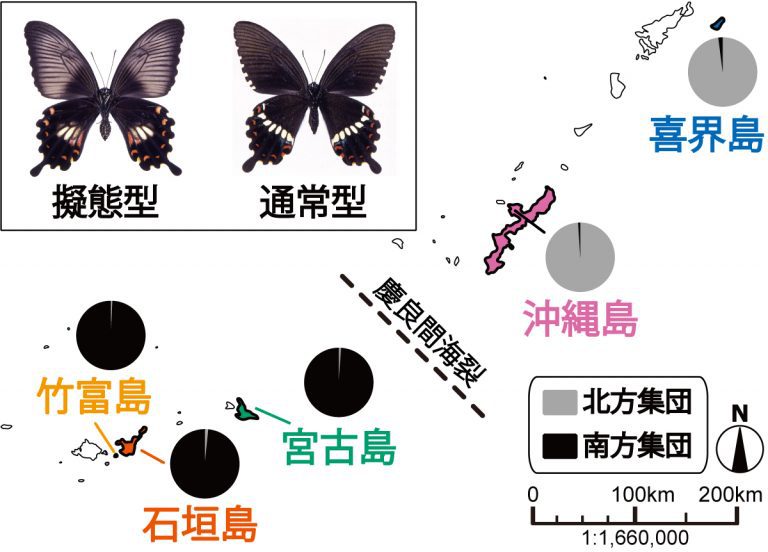
琉球大学・戦略的研究プロジェクトセンターの佐藤行人・特命講師、同・鶴井(佐藤)香織・特命助教と、同大学 農学部の辻瑞樹・教授を中心とする研究チームが、琉球列島に生息するシロオビアゲハ(図1; 計93個体; 調査地は喜界島、沖縄島、宮古島、石垣島、竹富島の5島)について、次世代シーケンサーを用いた遺伝子解析に基づいて詳細な島間・個体間の類縁関係を明らかにしました。
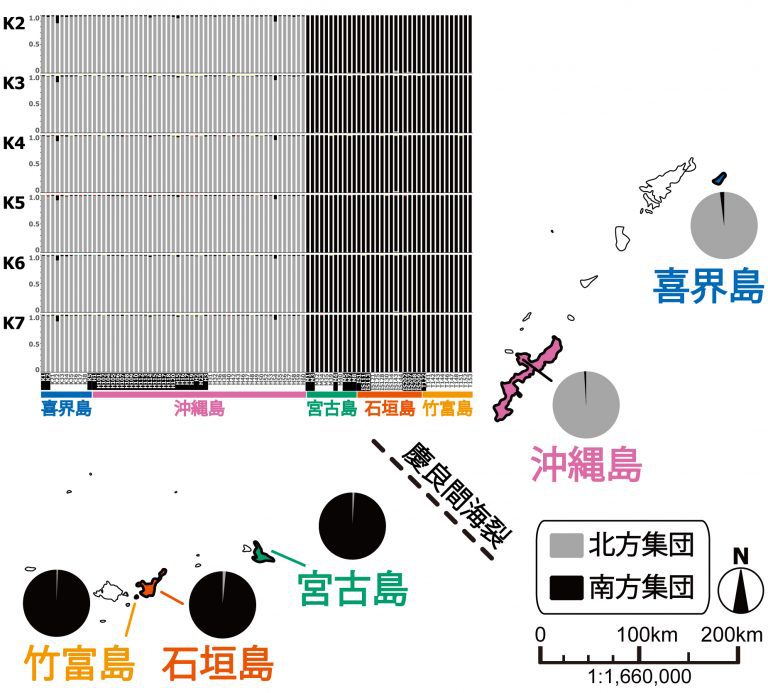
その結果、琉球列島のシロオビアゲハは、慶良間海裂の近辺を境界として、北方集団(喜界、沖縄島)と南方集団(宮古、石垣、竹富島)に、遺伝的に明瞭に分かれていること(図2)、南方集団は遺伝的多様性が高く比較的遠い過去から存続してきたと考えられる一方、北方集団は個体間の類縁性が高く、比較的近い過去に南方から移住してきたと考えられることを示しました。
さらに、今回の研究で93個体の遺伝子解析に使用できた238個のSNP(一塩基多型; 注1)の情報に基づいて、各島の間の平均遺伝距離を推定し、シロオビアゲハのメスの一部のみが示すベイツ型擬態の進化パターンについて解析を行いました。その結果、「各島でそれぞれ異なっている擬態型の存在割合は、各島に生息しているモデル種ベニモンアゲハ(図1左下)の個体数が多いほど高い」という、当研究チームが2019年に検証した仮説(Tsurui-Sato et al. 2019; 注2)が、本研究でも裏付けられました。琉球列島のシロオビアゲハに見られるベイツ型擬態の多様性は、各島の擬態モデル種の生息数、ひいては鳥類による捕食圧に対応して、各島で独自に進化してきたことが示唆されます。

<研究の背景>
シロオビアゲハ Papilio polytes は、東アジアの熱帯域から東南アジア、インド地方にまで分布するアゲハチョウ科の蝶であり、日本では九州南方の奄美大島より南に分布します。琉球列島では、この奄美大島、沖縄島から八重山諸島を含む多くの島々に生息し、生息数も多い普通種です。幼虫はシークヮーサーやサルカケミカン、ハマセンダンなどのミカン科樹木を食草とし、成虫(蝶)はハイビスカスの赤い花や、センダングサ(タチアワユキセンダングサ)の白い花に訪花する姿がよく見られます。
この、沖縄地方の普通種シロオビアゲハは、実は、進化論で有名なダーウィンの時代以来の謎を秘めた蝶でもあります。そのため、世界的にも多くの進化生態学・分子生物学的研究が行われてきました。その謎とは、『擬態する個体としない個体の混在』です。毒を持たないシロオビアゲハは、インドや台湾、タイ、そして琉球列島に渡る多くの生息地で、同じ地域に生息する有毒な蝶(毒蝶)の姿に似た姿へと進化する「ベイツ型擬態」を示す(図1)という特徴を持ちます(琉球列島では毒蝶ベニモンアゲハに擬態)。このベイツ型擬態によって、シロオビアゲハが捕食者の鳥類から食べられることを回避できていると考えられます。同じようなアゲハチョウ科のベイツ型擬態は、台湾のナガサキアゲハ(オオベニモンアゲハなどに擬態)や、北米東部のトラフアゲハ(アオジャコウアゲハに擬態)などにも見られます。
ダーウィンの自然選択の理屈で考えれば、ベイツ型擬態を示す個体は生存に有利なので、遅かれ早かれシロオビアゲハの全個体が擬態型となるはずです。しかし実際には、擬態はメスの一部でしか観察されません(図1)。この「生存に有利なはずの擬態型が、一部の個体でしか観察されない(擬態の多型)」という現象は、単純な自然選択の進化論では説明が困難です(図3)。ダーウィン自身は直接擬態の研究は行っていませんが、当時から擬態の不思議に関する議論は行われており、この擬態多型を説明するさまざまな仮説や理論が提案されてきました。代表的なものは、異性からの好みによって擬態模様が発達したという説(性選択)、擬態を発現する遺伝子や擬態型に不利な効果があるため生存率を下げるという説(擬態のコスト・トレードオフ; 注3)、擬態のモデル種(毒蝶)の生息数と比べて擬態個体が増え過ぎると、捕食者から見破られることで捕食され数が減るという説(頻度依存選択; 注2)などです(Barrett 1976; 上杉 2000; 大崎 2009)。我々はこれらの仮説のうち、主に擬態のコストと頻度依存選択に注目し、シロオビアゲハを対象に擬態型個体の比率の違いがどのような過程で生じるのかを探ってきました(Katoh et al. 2017; Tsurui-Sato et al. 2019; Katoh et al. 2020)。

<本研究のポイント>
我々は2019年に発表した論文(Tsurui-Sato et al. 2019; 注2)で、琉球列島のシロオビアゲハにおける「ベイツ型擬態の頻度依存選択説」について、フィールド調査とミトコンドリアDNA(注4)の遺伝子分析に基づいた検証を行いました。それによると、琉球列島の5島(喜界島、沖縄島、宮古島、石垣島、竹富島)に生息するベニモンアゲハの数が多いほど、シロオビアゲハの擬態型個体の割合(擬態率)が増加すること、また、各島に見られる擬態率の分布は、遺伝的類縁性や地理的距離、生息環境差では説明できないことから、頻度依存選択説が支持される結果となりました。
ミトコンドリアDNAを用いた遺伝子解析には、幾つか問題があります。まず、ミトコンドリアDNAの遺伝情報がシロオビアゲハの核遺伝子を含めた全遺伝情報のごく一部しか代表しないこと(本種のミトコンドリアDNA塩基数は約15 Kb、細胞核DNAの塩基数は約200~380 Mb; Gregory, T.R.ら、Animal Genome Size Databaseに基づき推定)、ミトコンドリアDNAは母親からしか子孫に伝わらないため、オスの遺伝情報が得られないことなどの不十分な側面があります。そのため上記研究の遺伝解析面は、父母の両方から受け継がれる細胞核DNAの遺伝子解析による裏付けが必要となります。そこで本研究はMIG-seq(注5)という手法を用いて、前の研究と同じ琉球列島5島のシロオビアゲハについて、細胞核DNAの遺伝子型を多数取得・分析し、詳細かつ高解像度な集団遺伝解析を行いました。
その結果、琉球列島に生息するシロオビアゲハは、慶良間海裂(図2)の近辺を境界に、北方集団と南方集団に明瞭に分かれるという集団構造を持つことが確認されました(図2、図4)。今回調べることができた5島は、喜界・沖縄島が北方集団、宮古・石垣・竹富島が南方集団に属します。
また、これら南北の集団の間で遺伝的多様性を比較すると、南方集団の平均対立遺伝子多様度(注6)は0.07~0.10と比較的高い一方、北方集団は0.04~0.06と比較的低いことが分かりました。南方集団が比較的遠い過去から存続してきたのに対して、北方集団はおそらく北上する下層ジェット気流に乗って、比較的近い過去に南方から移住してきた可能性が考えられます。このような亜熱帯域でのジェット気流に乗った島間移住は、ウンカ(Sogatella furcifera)などの農業害虫でも報告されています。とくに、北方集団の沖縄島のシロオビアゲハはヘテロ接合度(注7)が低く(0.08; 他の4島は0.20~0.27)、個体間の類縁度が高いと考えられることも、沖縄島の個体群が少数の祖先に由来することを示唆しており、上記の移住説を支持します(ただし、沖縄島集団に特異的な低頻度の変異が多型SNP座の約半数を占めることから、移住後ある程度の時間は経過していると推測される)。この琉球列島のシロオビアゲハに認められる南北の集団の存在と、南方集団の遺伝的多様性の高さ、南から北への移住の可能性については、ミトコンドリアDNA分析による研究(Tsurui-Sato et al. 2019; 注2)でも指摘されていました。今回の多数の細胞核SNPの解析から、移住説が改めて支持されました。
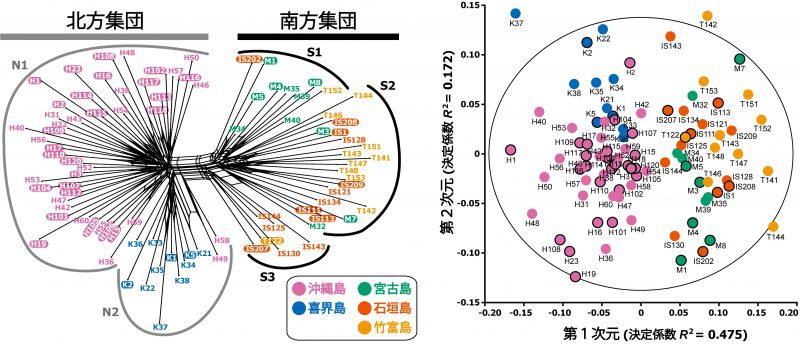
左側は遺伝距離(Kimuraの2パラメータモデル)に基づくネットワーク樹、右側は同遺伝距離に基づいた非計量多次元尺度構成法(nMDS)によるプロット。桃色は沖縄、青色は喜界、緑色は宮古、橙色は石垣、黄色は竹富の個体を表す。左のネットワーク樹でも、図2に示した集団判別分析の結果と同様に、沖縄・喜界の個体から成る北方集団と、宮古・石垣・竹富の個体から成る南方集団に明瞭に分かれる。白抜き文字で示した個体は擬態型であり、これらは単一のクラスターを形成しない。右側のプロットは、個体間の遺伝距離から求められる全個体ペア間の相互関係を2次元平面に投影した図で、黒枠の付いた個体は擬態型。縦横各軸の決定係数R 2は、データ全体に対するその軸の説明力を表す(最大1.0)。他の解析結果と同様に、喜界・沖縄の北方集団と、宮古・石垣・竹富の南方集団に大別され、両者の分布は重なり合わない。また、擬態型が特定の箇所に集中する傾向も見られない。
今回の細胞核SNPの分析によって、新たに判明した集団遺伝学的知見もあります。推定された遺伝距離に基づいて、各島集団を単位とした遺伝的類縁性を推定すると(図5)、個体レベルでの分析結果と同じく(図2、図4、図6)、宮古島は南方集団に属します。ミトコンドリアDNAに基づいた分析では、宮古島は喜界・沖縄島と共に北方集団に属していました(Tsurui-Sato et al. 2019; 注2)。今回の研究での核遺伝子情報に基づいた解析によれば、宮古島個体群は南方集団に属するものと解釈されます。今回明らかになったこのシロオビアゲハの生物地理区分は、他の生物群(トカゲ類など)の分布境界として知られる「慶良間海裂」とも整合します(図2)。さらに、ミトコンドリアDNAでは検出されなかった「距離による隔離」(地理的距離と遺伝的距離の相関関係)の効果が検出されました(細胞核SNPでの相関係数は0.90; ミトコンドリアDNAでは0.26)。このことは、近い場所に生息するチョウは遺伝的にも類似し、遠くなるほど遺伝的類似性が低下することを示しており、多数のSNP解析でより精度の高い集団遺伝情報が取得できたことを示します。
上記のように細胞核のSNP解析から推定された、より精度の高い島間の遺伝距離に基づいて、改めて「ベイツ型擬態の頻度依存選択説」を検討する解析を行ったところ、同説を裏付ける結果が得られました。調査地5島(喜界、沖縄、宮古、石垣、竹富島)のシロオビアゲハで観察された擬態率と、擬態型のモデル種である毒蝶ベニモンアゲハの生息数との相対頻度(毒蝶指数; 注2)の関係を統計的に解析したところ(偏マンテル検定)、偏相関係数が0.76~0.93と有意な高い値を示しました。前研究で推定された、ミトコンドリアDNAの遺伝距離に基づく同値は0.70~0.90であったことから(Tsurui-Sato et al. 2019; 注2)、今回のSNP解析によって、擬態型の頻度依存選択説はより強く支持されたことになります。
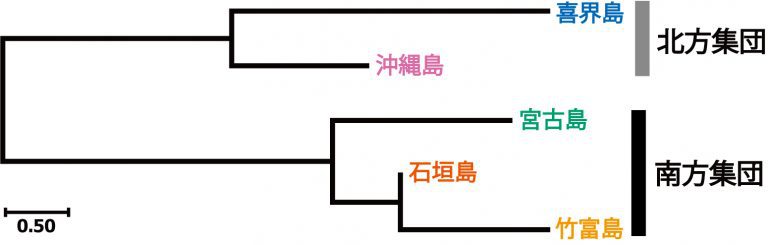
各島間の平均遺伝距離(Neiの純遺伝距離)に基づき近隣結合法により推定された島集団の間の遺伝的関係を表す。ミトコンドリアDNAに基づく分析では、宮古島集団は沖縄・喜界と同じ北方集団に含まれると推定されたが(Tsurui-Sato et al. 2019; 注2)、今回の細胞核SNP情報に基づいた、多数の遺伝子座の平均としては、宮古島集団は石垣・竹富とともに南方集団に属すると推定された。
<まとめと展望>
本研究では、MIG-seq(注5)という細胞核のSNPを解析する手法を用いて、琉球列島に生息するシロオビアゲハの詳細な集団遺伝構造を推定し、それにより実現した精度の高い遺伝距離などの解析に基づいて、我々が焦点を当ててきたベイツ型擬態の頻度依存選択説を検証することができました。
今回採用した細胞核SNPの解析手法MIG-seq(注5)の利点は、比較的少量のDNAサンプルしか手に入らない野生生物にも適用できることです。それは、PCR(注8)によってSNPを含むDNA領域を増幅して解析するためです。一方、野生生物サンプルから手に入るDNAサンプルの質や実験の設定によっては、遺伝子増幅の際に特定のSNP領域が多く増幅されてしまい(PCRバイアス)、結果として利用できるSNPの数が少なくなる場合もあります。例えば今回のシロオビアゲハでは、個体あたり平均4,864個のSNP座が取得されましたが、93個体間の比較解析に使用できた欠損値の少ない(<20%)SNP座は、238個まで減少しました。このSNP数は、ある程度の集団遺伝学解析を行うことは可能ですが、連鎖地図推定や遺伝子探索といった目的には不十分です。
上記より、琉球列島のシロオビアゲハ研究における有望な展開の1つは、集団レベルで全ゲノムを解析することであると考えられます。近年、ゲノム規模のDNA配列決定を行う次世代シークエンサー(注9)のコストパフォーマンスが向上してきました。シロオビアゲハのドラフトゲノム配列(注10)の構築と、集団レベルでの全ゲノムSNP解析を行うことができれば、より詳細な集団遺伝構造や個体間の関係(図6)が解明されてきます。すると、アゲハチョウ属 Papilio の擬態形質の発現に関係すると考えられるdsx(doublesex)遺伝子の集団遺伝動態や、擬態型シロオビアゲハの斑紋形成に関わる未知の遺伝子探索などが進むと期待されます。擬態の生理・分子メカニズムと集団ゲノミクスの研究を併せて進めることによって、琉球列島のシロオビアゲハに代表されるベイツ型擬態の総合的な進化生態研究が、さらに進展するものと期待されます。

各個体が、互いにどの段階で共通祖先に辿ると推定されるかを、確率分布で示した系統樹。遠い祖先へ遡るほど推定の分散が増大するため、推定像も曖昧となる。
<用語解説>
(注1)SNP: single nucleotide polymorphism(一塩基多型)の略。相同なDNA配列領域(遺伝子座)について、同種の個体間や、同一個体内の対立遺伝子間で配列を比較した時に、DNAの文字(塩基)が異なっている部位または状態のこと。多くの場合、共通祖先のいずれかの段階で起きた突然変異(塩基置換)に由来しており、遺伝的多様性や系統関係などの集団遺伝学・分子系統学的分析を行う情報源として利用される。大多数のSNPは、生存に有利でも不利でもない(進化的に中立)と考えられるが、一部には疾患の原因となるものや、致死性のもの、生物機能を変えるものも存在し、長期的には生物進化の材料の一部である(挿入・欠失、遺伝子重複、転移因子やウィルス配列の挿入など、他の種類の突然変異も存在する)。
(注2)琉球大学2019年4月25日プレスリリース: 「シロオビアゲハが毒蝶を真似するときの条件 ~擬態の進化学的パラドクスを解明~」
(注3)琉球大学2020年8月26日プレスリリース: 「蝶の成虫に役にたつ“擬態遺伝子”が幼虫や蛹では生存率を下げる?」
(注4)ミトコンドリアDNA: 真核生物の細胞内小器官ミトコンドリア内にあるDNA。ミトコンドリア内膜上で機能するタンパク質の一部と、それらを合成するリボソーム(遺伝情報に基づいてタンパク質を合成するタンパク質–RNA複合体)を構成するRNAの一部、およびミトコンドリア内で使用されるtRNA(タンパク質合成時にアミノ酸を運搬するRNA)をコードする。ミトコンドリアDNAは、一細胞に多数(数百から数千分子)含まれており、比較的容易に分析できる。そのため、塩基配列情報が乏しい野生生物で研究に活用されることが多い。
(注5)MIG-seq: multiplexed inter-simple sequence repeat genotyping by sequencing(単純反復配列間領域の並列化配列決定による遺伝子型解析)の略。真核生物の細胞核ゲノム中に散在する単純反復配列(CTACTA、TGTGTGなど)をターゲットとして、ポリメラーゼ連鎖反応(PCR; 注8)によるDNA増幅を行い、増幅された反復配列に挟まれる領域のDNA配列を、次世代シークエンサー(注9)で大量に解読する手法。得られた配列情報をサンプル個体間で比較することにより、ゲノム内に存在するSNP(一塩基多型)の情報を取得し様々な解析に用いることを目的とする。
(注6)平均対立遺伝子多様度(average gene diversity): 調べた遺伝子座について、ランダムに抽出した対立遺伝子が異なったものである(ヘテロである)確率。全ゲノム、または総塩基長が分かっているDNA配列の塩基多様度πとは異なる尺度であり、当該研究で調べられた遺伝子座やSNP(注1)の情報のみから推定される値である。
(注7)ヘテロ接合度(H o, observed heterozygosity): ある生物集団の相同な遺伝子座を調べた時に、個体内の対立遺伝子が異なるものであった確率。集団遺伝学において、集団の遺伝的多様性を表す尺度の1つとして使われる。
(注8)PCR: polymerase chain reaction(ポリメラーゼ連鎖反応)の略で、DNAの集合から、ターゲットとする特定の配列に挟まれた領域を生化学的に増幅する手法。微量のDNAサンプルから、多数のコピーDNAを得て、配列決定などの分析に使用することができる。
(注9)次世代シークエンサー: 数千万から数億分子のDNAについて、同時並列的に塩基配列を解読することができる機器。2004年ごろから普及し、現在はヒトゲノムの解読や微生物叢の研究をはじめとして、生物学で使われる重要技術の1つになっている。
(注10)ドラフトゲノム配列: ある生物の全遺伝情報(ゲノム)の概要配列情報のこと。ゲノムを構成するDNA塩基の大部分ないし主要な多くの部分を解読し、コンピュータ解析によってつなぎ合わせたDNA配列の集合。概要と呼ぶのは、DNA配列を決定することが、実験上またはデータ解析上困難な反復配列領域やテロメア領域など、解読できていないゲノム領域が残るためである。このようにドラフトゲノム配列は、一般に全遺伝情報を必ずしもカバーしていない場合が多い。また、つなぎ合わせたDNA配列について、染色体上での相対的な位置関係(連鎖地図)や、核型や染色体上の絶対位置との対応関係(物理地図)が明らかにされているものと、明らかにされていない配列情報のみのものとがある。
<引用文献>
Barrett, J. A. (1976). The maintenance of non-mimetic forms in a dimorphic Batesian mimic species. Evolution, 30, 82‒85.
上杉兼司 (2000). 成虫はどうやって身を守っているのか. 擬態と多型. 『蝶の自然史 行動と生態の進化学』(大崎直太・編著), 北海道大学図書刊行会, pp. 106‒123.
大崎直太 (2009). 『擬態の進化. ダーウィンも誤解した150 年の謎を解く』, 海游舎.
Katoh, M., Tatsuta, H., & Tsuji, K. (2017) Rapid evolution of a Batesian mimicry trait in a butterfly responding to arrival of a new model. Scientific Reports, 7, 6369.
Tsurui-Sato, K., Sato, Y., Kato, E., Katoh, M., Kimura, R., Tatsuta, H., & Tsuji, K. (2019). Evidence for frequency-dependent selection maintaining polymorphism in the Batesian mimic Papilio polytes in multiple islands in the Ryukyus, Japan. Ecology and Evolution, 9, 5991‒6002.
Katoh, M., Tatsuta, H., & Tsuji, K. (2020). Mimicry genes reduce pre-adult survival rate in Papilio polytes : a possible new mechanism for maintaining female-limited polymorphism in Batesian mimicry. Journal of Evolutionary Biology, 33, 1487‒1494.
<謝辞>
当研究チームに、琉球大学農学部の卒業研究生として参加した加藤絵美氏の寄与に感謝申し上げます。シロオビアゲハとベニモンアゲハの標本写真は、野林千枝氏からご提供賜りました。本研究は、琉球大学 研究プロジェクト推進経費(戦略的研究推進経費 No.16SP01302)の助成のもとで行われました。また、同大学・時空間ゲノミクスプロジェクト、研究基盤センター、ならびに戦略的研究プロジェクトセンターによる共同利用・共同研究の支援を受けました。また、情報・システム研究機構・国立遺伝学研究所が有する遺伝研スーパーコンピュータシステムを利用しました。
<本記事内の図およびイラストレーション>
本記事で使用した図とイラストレーションは、当該論文(下記)の著者らによって作成されました。研究結果を説明する図は、当該論文が掲載している図(figures)に準拠して、本記事向けにレイアウトの変更や日本語への翻訳を施して作成しました。イラストレーションは、生物の写真や肖像写真に基づいて、主著者・佐藤が作成しました。
<論文情報>
論文タイトル: Population genetic structure and evolution of Batesian mimicry in Papilio polytes from the Ryukyu Islands, Japan, analyzed by genotyping-by-sequencing(和訳)DNA配列決定による遺伝子型解析に基づいた琉球列島におけるシロオビアゲハ Papilio polytesの集団遺伝構造とベイツ型擬態の進化
雑誌名: Ecology and Evolution
著 者: Yukuto Sato, Kaori Tsurui-Sato, Mitsuho Katoh, Ryosuke Kimura, Haruki Tatsuta, Kazuki Tsuji
佐藤行人(琉球大 戦略的研究プロジェクトセンター 特命講師)、鶴井(佐藤)香織(琉球大 戦略的研究プロジェクトセンター 特命助教)、加藤三歩(琉球大 農学部および鹿児島大院 連合農学研究科 研究員)、木村亮介(琉球大院 医学研究科 准教授)、立田晴記(琉球大 農学部および鹿児島大院 連合農学研究科 教授)、辻瑞樹(琉球大 農学部および鹿児島大院 連合農学研究科 教授)
DOI番号: 10.1002/ece3.7092
URL: https://doi.org/10.1002/ece3.7092



-アイキャッチ-520x520.jpg)